
Theoretical chemistry compliments laboratory-based research by investigating the fundamental principles underlying chemical processes. Computational methods are valuable tools for predicting a wide range of chemical properties, including thermochemistry, reaction mechanisms, chemical kinetics and spectroscopic quantities, allowing high-throughput screening of compounds. Our faculty develop new theoretical methods and use high performance computing to investigate important problems in molecular, biological, and materials chemistry.
Ph.D. students choose from graduate classes in Molecular Quantum Chemistry, Statistical Thermodynamics, Electronic Structure Theory, Condensed Matter Physics, Molecular Dynamics and Biomolecular Simulation, Molecular Spectroscopy, Density Functional Theory, Numerical Mathematical Methods, Deep Learning, Scientific Computing.
Theoretical chemistry grad students often learn together in our student run journal club, organizing workshops on various topics such as python, Jupyter notebooks, Fortran, Hartree-Fock theory, and machine learning. We often have annual retreats with presentations, lightening talks, tutorials, Q&A panels, and community building fun.
More about our faculty and their research groups:
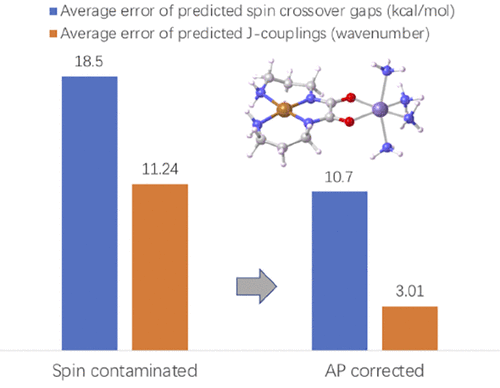
The Hra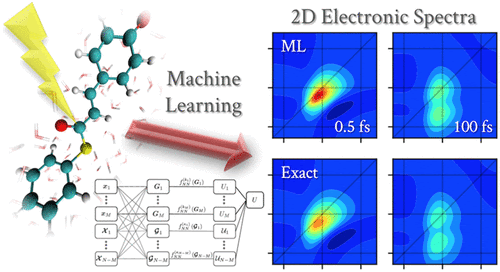 tchian group develops and applies new computational models for studying chemical reactivity, especially in the area of transition metal catalysis.
tchian group develops and applies new computational models for studying chemical reactivity, especially in the area of transition metal catalysis.
The Isborn group is developing and improving existing quantum mechanical and classical methodologies to more accurately model solvation, electronic excitation, charge transfer, and nonlinear spectroscopy.
The Larsson group focuses on developing and utilizing tools for simulating and understanding quantum effects, quantum dynamics, and strong correlation in molecular systems.
The Pribram-Jones group analyzes temperature effects, time dependence, and ensemble behavior in existing and new methods for simulating atomic excitations, high entropy alloys, and warm dense matter.
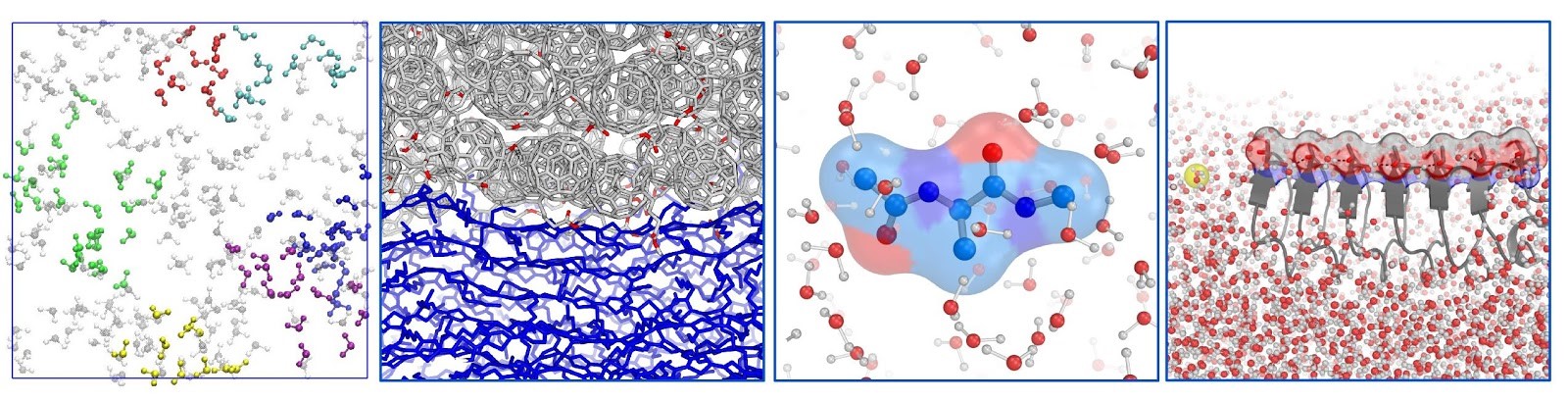
 The Shi group develops and applies multi-scale modeling methods to understand the structure, dynamics and spectroscopy of complex condensed-phase molecular systems.
The Shi group develops and applies multi-scale modeling methods to understand the structure, dynamics and spectroscopy of complex condensed-phase molecular systems.
The Strubbe group develops and applies methods for excited-state dynamics in molecular, nanoscale, and solid-state systems.


